The polyol pathway is a two step process that converts glucose to fructose. This post explores how copper deficiency may make intermediates in this pathway worse.These findings have relevance to Alzheimer’s Disease.
Xu J, Begley P, Church SJ, Patassini S, McHarg S, Kureishy N, Hollywood KA, Waldvogel HJ, Liu H, Zhang S, Lin W, Herholz K, Turner C, Synek BJ, Curtis MA, Rivers-Auty J, Lawrence CB, Kellett KA, Hooper NM, Vardy ER, Wu D, Unwin RD, Faull RL, Dowsey AW, Cooper GJ. Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer’s disease: metabolic basis for dementia. Sci Rep. 2016 Jun 9;6:27524. PMC free article
Introduction …. to Alzheimer’s Disease
“Alzheimer’s disease†(AD) may occur through interplay of environmental, genetic, and metabolic factors according to these authors. The authors list some pathological manifestations:
- Aβ-amyloid deposition
- neurofibrillary tangles comprising tau protein
- impaired cerebral glucose metabolism (‘hypo metabolism’) with insulin resistance and defective carbohydrate regulation
- oxidative stress and inflammation
- cerebrovascular amyloid angiopathy (CAA)
- enhanced advanced glycation end-product (AGE) formation
- defective copper regulation.
- defects in glucose uptake
The polyol pathway
The polyol pathway is a means of taking the six carbon sugar glucose with the aldehyde C=O group on the top of the structure to a ketone form, fructose, with the C=O on the second carbon from the top. In this way, the propensity to form advanced glycation end products is reduced.
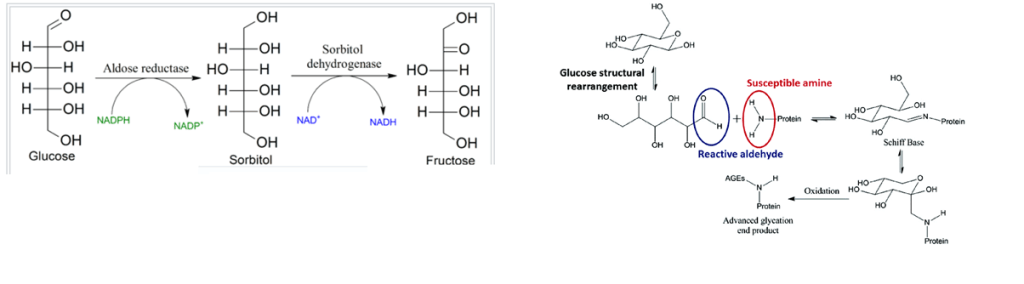
Copper should theoretically increase the utilization of glucose if copper is a pivotal player in brain glucose utilization simply by keeping the electron transport chain running.
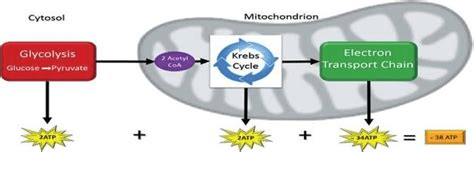
Our brains really need ATP. Copper is an important part of the electron transport chain that maximizes the yield of ATP from glucose. It makes sense that copper deficiency might lead to a pile up of glucose that then gets shunted to sorbitol and fructose.
The authors analyzed post-mortem tissue from seven brain regions of patients with AD and controls who did not have clinical evidence of dementia. The possibility exists that one or more patients had undiagnosed mild T2D or pre-diabetes. The authors reported that elevated glucose levels have previously been linked to altered tissue-copper regulation in the context of diabetes.
Regions most prone to damage in AD
- hippocampus
- entorhinal cortex
- middle temporal gyrus
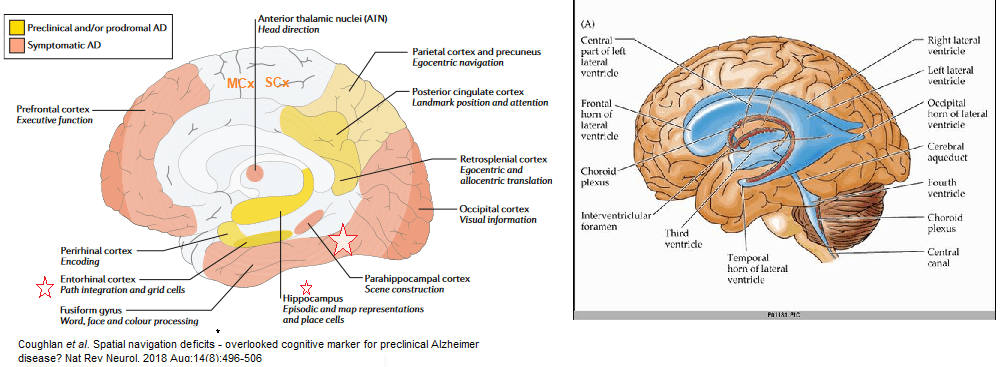
Three regions are less effected
- cingulate gyrus,
- sensory cortex
- motor cortex
The cerebellum is largely spared.
The inverse relationship between glucose and copper
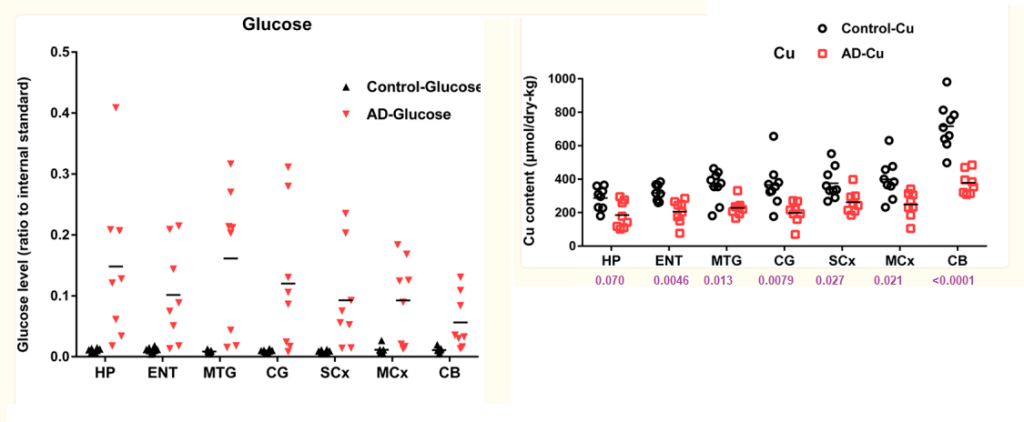
Free glucose is elevated in AD brain, particularly in vulnerable regions where elevated glucose may well be linked to copper deficiency and tissue-damage.
- Overall mean brain-glucose levels were higher (P = 9.7 × 10−13) in AD vs controls
- Brain copper values lower (P = 1.6 × 10−8) in cases vs controls.
P values that the AD is different from the controls is written below the data points. There was modest evidence that overall brain-copper and brain-glucose values were inversely correlated (P = 0.021) in AD-patients but not controls
Brain-sorbitol and brain-fructose levels are also elevated in AD brain, consistent with increased polyol pathway flux. Defects may be in glucose utilization, the TCA cycle, or the electron transport chain.
The six carbon sugar pile up in AD and T2D
A further, mechanistically-related finding is that pan-cerebral brain-copper deficiency accompanies these glucose and polyol-pathway defects: elevated tissue-CML levels provide a molecular linkage between elevated brain glucose and copper deficiency.
Impaired neuronal glucose utilization could play a major role in tissue-damage in AD, possibly via defective mitochondrial metabolism caused by deficient copper supply to cytochrome C oxidase subunits I and II (COI and COII), leading to impaired function of cytochrome c oxidase. These findings provide a clear molecular linkage between mechanisms of tissue damage in AD and T2D.
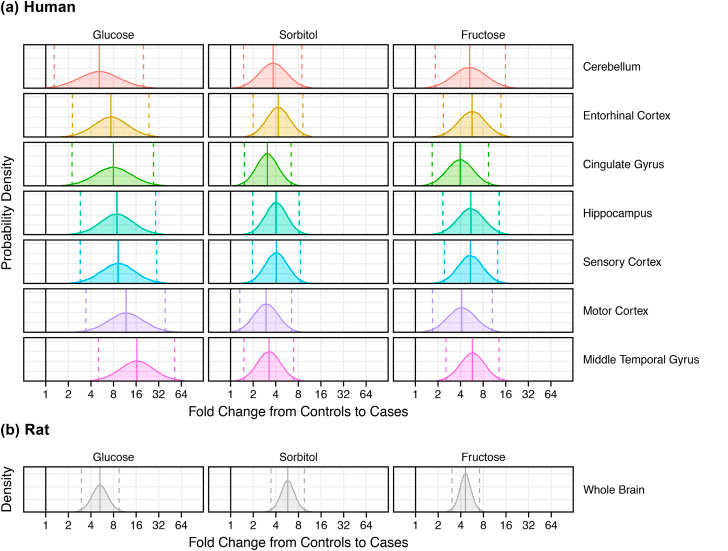
These “fold change” plots compare controls and disease in tissue from (a) each of seven brain regions from patients with AD (n = 9) and controls (n = 9) and (b) whole brain homogenates from diabetic (n = 7) and control (n = 7) rats.
- Complicated methods of analysis are not presented in this post.
- Solid lines are average fold changes.
- Dashed lines are the 95% Confidence intervals.
- The dark line at “1” represents a fold change of 1, that is “no change”
The process by which elevated brain glucose is linked to defective cell-copper uptake and intracellular transport to key copper proteins such as COI, COII, SOD1 and SOD3, may well provide a new target for diagnostic imaging or therapeutic intervention.
A few words of extreme caution
The same authors performed a followup study in post mortem brains of type 2 diabetes patients. Transition metal levels were measured, but glucose, sorbitol, and fructose were not. In contrast to AD patients, copper was found to be elevated in the hippocampus of T2D patients.
Philbert SA, Schönberger SJ, Xu J, Church SJ, Unwin RD, Cooper GJS. Elevated hippocampal copper in cases of type 2 diabetes. EBioMedicine. 2022 Dec;86:104317. PMC free article
This is a direct quote from the discussion of the data
“Despite copper’s key roles in some antioxidant processes (e.g., those catalysed by superoxide dismutase 1 and by complex IV), it can also participate in the production of harmful reactive oxygen species (ROS) via the Fenton and Haber–Weiss reactions. There is evidence that CuII can bind to advanced glycation end-products (AGEs) whilst retaining its redox–active properties. As protein glycation is significantly increased in T2D due to hyperglycaemia and polyol-pathway activation, the elevated hippocampal copper measured in the present study has the potential to cause increased AGE-CuII complex formation. The formation of AGE-CuII complexes could thereby lead to increased CuII-mediated ROS production, and decreased copper bioavailability for antioxidative pathways due to the selective binding of extracellular CuII with AGEs. However, CuII is less abundant than CuI, so the role that AGE-CuII complexes might play in the pathogenesis of T2D remains to be clarified.”
Stay tuned for more on these AGE-CuII complexes.
Leave a Reply